
Resumo sobre epidermólise bolhosa (completo): da definição ao tratamento, passando pela epidemiologia, diagnóstico e quadro clínico. Confira!
Definição
A epidermólise bolhosa (EB) é um grupo heterogêneo de desordens cutâneas geneticamente transmitidas, que se caracteriza pela formação de bolhas espontâneas ou após trauma local.
Existem quatro tipos principais de EB: EB simples, EB juncional, EB distrófica e síndrome de Kindler, diferenciados pelo nível de clivagem das bolhas e subdivididos de acordo com o padrão de herança genética, morfologia/topografia das lesões e mutação genética envolvida.
Epidemiologia da epidermólise bolhosa
EB hereditária é de ocorrência mundial e acomete ambos os sexos. Não há dados epidemiológicos sobre sua frequência no Brasil. De acordo com a literatura, a prevalência de EB fica em torno de 11 casos por um milhão de habitantes e a incidência de aproximadamente 20 casos por um milhão de nascidos vivos.
A taxa de incidência de EB, por subtipo, é aproximadamente oito por milhão de nascidos vivos para EB simples, três por milhão de nascidos vivos para EB juncional, dois por milhão de nascidos vivos para EB distrófica dominante e três por milhão de nascidos vivos por EB distrófica recessiva.
Patogênese da epidermólise bolhosa
A epidermólise bolhosa simples (EBS) é o tipo mais comum de EB e é causada por mutações dominantes autosomal nas KRT5 e KRT14, genes que codificam as queratinas, resultando em clivagem do tecido ao nível dos queratinócitos basais.
A epidermólise bolhosa juncional (EBJ) é causada por mutações autossômicas recessivas nos genes LAMA3 , LAMB3 e LAMC2 que codificam a laminina-332, resultando na clivagem do tecido na lâmina lúcida do BMZ. EBJ intermediário generalizado (EBJ-não Herlitz) é causado por mutações que afetam predominantemente COL17A1 (que codifica colágeno tipo XVII) e mostra um curso geralmente mais suave.
A epidermólise bolhosa distrófica (EBD) é causada por mutações autossômicas dominantes ou recessivas no gene COL7A1 que codifica a cadeia alfa-1 do colágeno tipo VII, resultando na clivagem do tecido abaixo da lâmina densa do BMZ.
A síndrome de Kindler (SK) é um tipo de EB autossômica recessiva causado por mutações no gene FERMT1 , que codifica a proteína de adesão focal homóloga 1 da família da fermitina, também chamada de kindlin-1, resultando na clivagem do tecido em níveis variáveis na zona BMZ.
Manifestações clínicas
Epidermólise bolhosa simples: é caracterizada por formação de bolha intraepidérmica e pouco envolvimento sistêmico. Distrofia ungueal, alopecia e lesões em mucosas podem ocorrer nas formas mais graves. As lesões cutâneas geralmente desaparecem sem deixar cicatrizes. A formação de bolhas diminui com o aumento da idade.
Epidermólise bolhosa juncional (EBJ): o envolvimento da mucosa oral, alopecia e anoníquia são frequentes. Há descritos na literatura casos de EBJ com atresia congênita do piloro e, mais raramente, de outras porções do trato gastrointestinal. Esta desordem está associada a risco significativo de anomalias congênitas do trato geniturinário e morte infantil/neonatal. A EBJ generalizada grave se apresenta com vesículas mucocutâneas extensas ao nascimento e está associada à letalidade precoce.
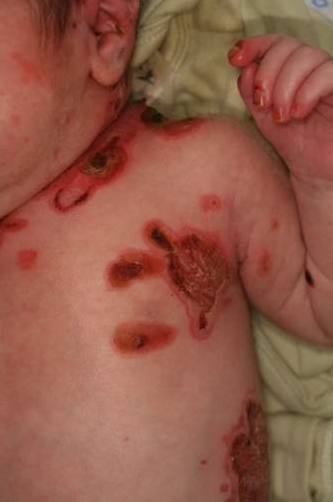
Epidermólise bolhosa distrófica (EBD): as características clínicas da são fragilidade da pele, bolhas, cicatrizes, alterações nas unhas e formação de milia. Como o colágeno VII também é expresso em epitélios estratificados não cutâneos, a formação de bolhas também ocorre nas membranas mucosas e no terço superior do esôfago.
O espectro fenotípico varia desde o EBD dominante mais suave, em que os pacientes têm apenas unhas distróficas, até a distrófica recessiva grave generalizada, no qual há bolhas e cicatrizes generalizadas levando à pseudosindactilia (fusão dos dedos).

Síndrome de Kindler: É uma dermatose rara, caracterizada pela formação de bolhas acrais, fusão de quirodáctilos/ pododáctilos, além de poiquilodermia generalizada e progressiva. Outros achados clínicos incluem bolhas induzidas por trauma (comum a todas as EBs hereditárias), pele seca e atrófica, liquenificação e fotossensibilidade de superfícies proximais.
Manifestações extracutâneas: são comuns a todos os subtipos graves de EB e incluem anormalidades no cabelo e nas unhas, bolhas e cicatrizes intraorais e anomalias dentárias, estenoses esofágicas e anormalidades geniturinárias. Infecções cutâneas, sepse, desnutrição, anemia e carcinoma de células escamosas são complicações frequentes e estão associadas a morbidade e mortalidade significativas.
Diagnóstico da epidermólise bolhosa
Deve-se suspeitar de EB em indivíduos com história de bolhas ou erosões recorrentes após trauma leve e em neonatos que apresentam bolhas e erosões sem outra etiologia plausível (por exemplo, infecção), especialmente se a história familiar for positiva para manifestações semelhantes. A biópsia de pele para mapeamento de imunofluorescência (IFM) de uma bolha recentemente induzida é a abordagem de primeira linha para o diagnóstico de EB, na maioria dos casos.
Tratamento da epidermólise bolhosa
O tratamento não farmacológico consiste na proteção das lesões, prevenção de novas lesões, controle da dor e do exsudato. As opções disponíveis para o tratamento da Epidermólise Bolhosa referenciam-se à fibras de absorção do exsudato, espumas não aderentes, espumas com microaderência seletiva e aquelas com impregnação de antibacterianos e anti-inflamatórios, auxiliadores de reepitelização e malhas não aderentes.
Antibióticos tópicos ou sistêmicos podem ser utilizados por curtos períodos nos casos em que a ferida não cicatriza e que há aumento de exsudato, eritema, presença de tecido friável, presença de tecido morto e mau cheiro.
O controle da dor é um aspecto importante do manejo de pacientes com EB. Para dor leve a moderada, analgésicos como dipirona e paracetamol podem ser usados sozinhos ou em conjunto com um antiinflamatório não esteroidal. Para dores intensas, opióides ou ansiolíticos podem ser necessários.
O suporte nutricional desempenha um papel crítico na resolução das feridas. Alguns pacientes requerem um tubo de gastrostomia para otimização do status nutricional. Também é importante monitorar e manter os níveis de hemoglobina acima de 8mg/dl. Pacientes anêmicos requerem suplementação de ferro com ou sem eritropoietina. A suplementação de cálcio e vitamina D deve ser iniciada em pacientes com evidência de osteopenia ou osteoporose.








