
Confira as principais informações que você precisa saber a respeito da dermatomiosite!
A dermatomiosite (DM) é uma miopatia imunomediada que, junto com a polimiosite (PM), possui como características comuns: fraqueza do músculo esquelético proximal e evidência de inflamação não supurativa da musculatura estriada.
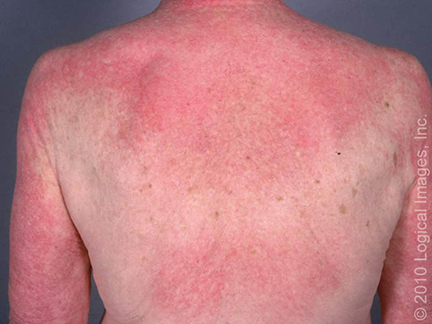
A DM, ao contrário da PM, está associada a uma variedade de manifestações cutâneas características. As lesões cutâneas características são heliótropo, pápulas de Gottron e eritrodermia generalizada.
É importante salientar que essa é uma doença sistêmica e acomete diversos órgãos. Existe ainda a forma de DM amiopática, que é uma condição muito mais rara, na qual os pacientes apresentam apenas achados cutâneos característicos, sem fraqueza ou enzimas musculares anormais.
Os diagnósticos diferenciais de dermatomiosite são diversos, incluindo doenças que não comprometem a pele, como a depressão maior.
Isso ocorre porque essa patologia faz parte de um grupo de doenças atualmente referidas na literatura como infecção de partes moles.
Veja abaixo o resumo que preparamos para te ajudar na identificação dessa doença!
Epidemiologia da dermatomiosite
Muitos estudos de prevalência analisam a dermatomiosite e polimiosite como patologias que ocorrem em associação, sendo difícil compreender a epidemiologia mais específica da DM.
A prevalência que é estimada atualmente é de 5 a 22 a cada 100.000 habitantes e a incidência é de 2 a 10 casos por milhão de habitantes.
Em um estudo de base populacional em Minnesota, nos Estados Unidos, analisaram-se dados de 1976 a 2007, onde a incidência anual estimada de todos os subtipos de DM foi de aproximadamente 1 por 100.000 pessoas.
Nesse mesmo estudo, a incidência anual estimada para o subtipo clinicamente amiopático de DM foi de 0,2 por 100.000 pessoas.
Em relação ao sexo biológico, compreende-se que as mulheres são duas vezes mais acometidas que os homens, mas na doença juvenil a incidência é igual entre os sexos.
Existe um pico bimodal de incidência da DM, sendo o primeiro em crianças entre os 10 e 15 anos de idade; e o segundo pico na faixa entre 40 e 50 anos.
Patogênese da dermatomoisite
Os mecanismos patogênicos específicos que são responsáveis pela dermatomiosite (DM) são desconhecidos. No entanto, acredita-se que sejam desencadeadas por fatores ambientais em indivíduos geneticamente suscetíveis.
As anormalidades moleculares sugerem fortemente uma patogênese incluindo a superprodução de um IFN tipo 1, com as evidências disponíveis sugerindo predominantemente IFN-beta (IFNB).
O papel patogênico dos autoanticorpos específicos da dermatomiosite e suas manifestações também é incerto.
Alguns estudos sugerem a associação com certos genes de HLA, como o HLA DQA1 0501 para DM juvenil e polimorfismo 308 do TNF-alfa em pacientes com fotossensibilidade na DM.
Outros relatos sugerem que alguns fatores ambientais podem estar relacionados com o aparecimento de MII, como toxinas, uso de estatinas, corticoide e D-penicilamina.
No geral, a dermatomiosite é considerada uma doença imunologicamente mediada e o alvo antigênico primário é o endotélio, com consequente ativação do sistema complemento.
Nela está presente a fração terminal do complemento C5b-9 de ataque à membrana nas paredes dos vasos, o que facilita a migração para o músculo de anticorpos ligados ao complemento, células B, T CD4+ e macrófagos.
Esse conjunto de ações levam à redução do número de capilares, hipoperfusão endofascicular e à alteração característica da DM, que é a atrofia perifascicular.
Autoanticorpos com afinidades por vários antígenos, como as sintetases do RNA de transferência de aminoacil (tRNA), foram relatados em DM e outras miopatias inflamatórias.
O papel patogênico de qualquer um desses autoanticorpos é incerto e continua a ser explorado, embora a detecção desses autoanticorpos possa ser útil clinicamente por causa de sua associação com síndromes clínicas específicas.
Quadro clínico da dermatomiosite
Alguns achados cutâneos são típicos da dermatomiosite (DM), como pápulas rosa-violáceas cobrindo as articulações interfalangianas e metacarpofalangianas (pápulas de Gottron).
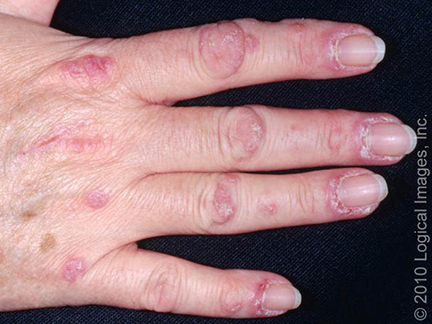
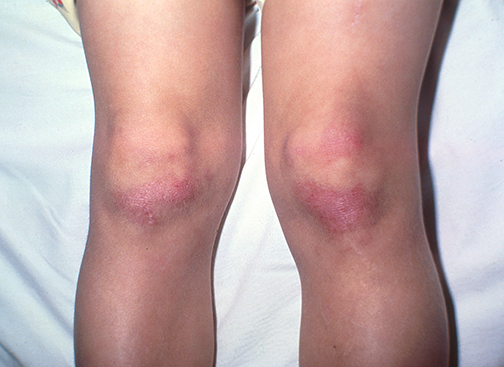
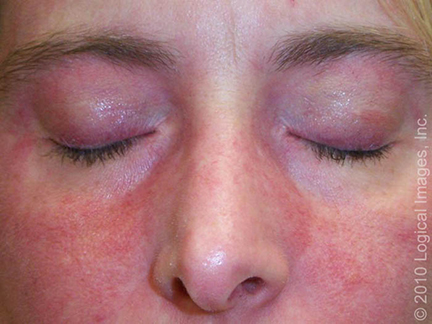
Podem surgir também um eritema macular rosa-violáceo sobre outras articulações, como cotovelos ou joelhos (sinal de Gottron), e a erupção do heliotrópio, um eritema rosa-violáceo, com ou sem edema, envolvendo a pele periorbital.
A deposição de cálcio na pele, um achado conhecido como calcinose cutânea, ocorre geralmente na DM juvenil, não sendo frequente na DM no adulto. Alguns casos apresentam regressão espontânea sem tratamento específico.
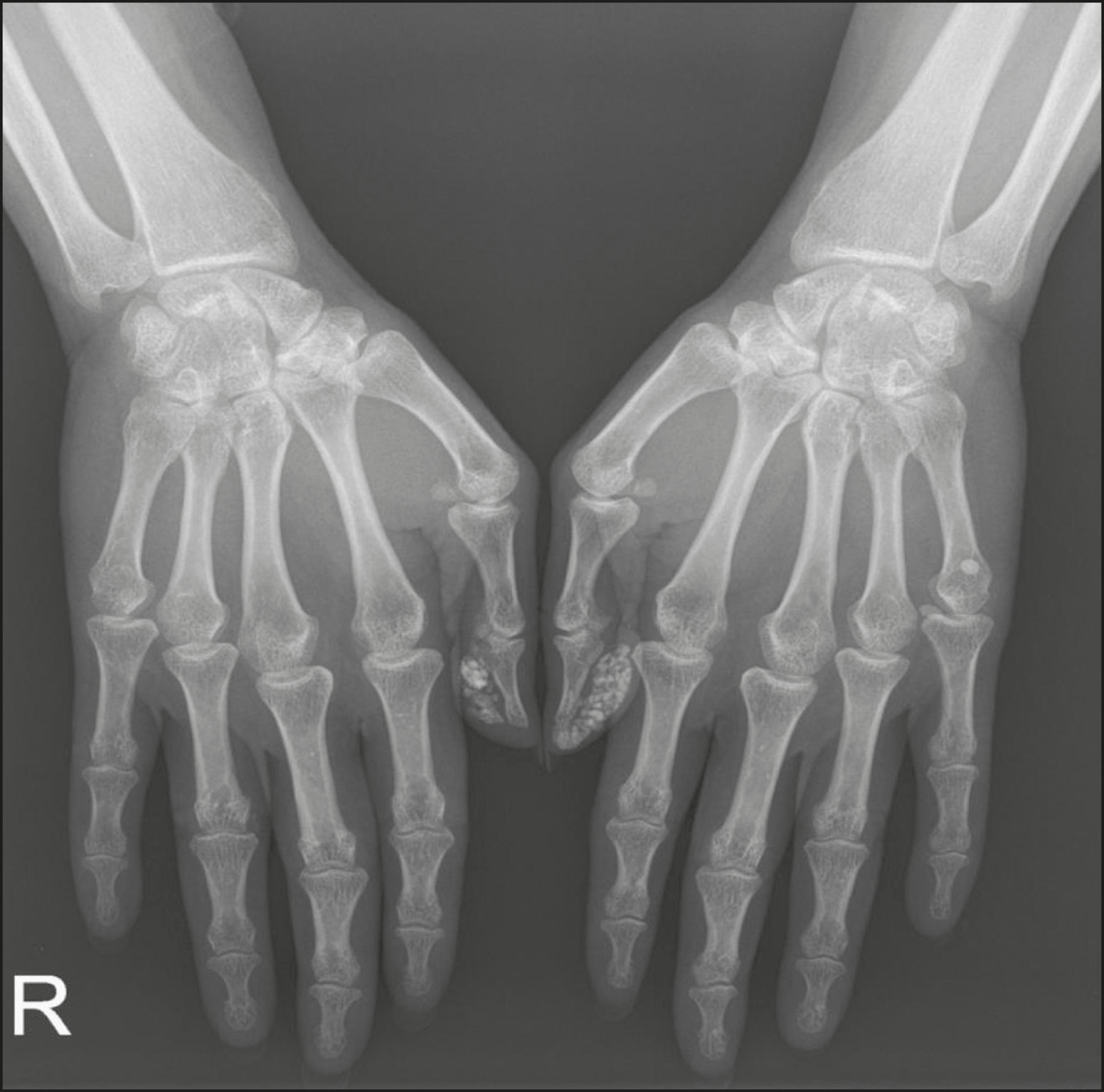
A maioria dos pacientes com DM apresenta acometimento cutâneo e muscular simultâneo, evidenciado por fraqueza muscular proximal e teste diagnóstico que revela a presença de miosite.
No entanto, o início da doença cutânea pode preceder o aparecimento da miosite em até vários meses em 30% dos pacientes com DM clássico e segue logo após o envolvimento muscular em 10%.
Alguns pacientes apresentam poliartrite simétrica de pequenas articulações, especialmente em fases precoces da doença.
A musculatura faríngea pode ser acometida, causando disfagia superior, manifestada como dificuldade de início da deglutição, regurgitação nasal ou disfonia.
O envolvimento pulmonar ocorre em até 50% dos pacientes. Pneumonia aspirativa, geralmente recorrente, é prevalente em pacientes com fraqueza em musculatura faríngea.
Doença pulmonar intersticial ocorre em mais de 30% dos casos e em aproximadamente 60% daqueles com anticorpos anti-sintetases (anti-Jo1).
Diagnóstico de dermatomiosite
O diagnóstico de dermatomiosite cutânea (DM) é sugerido pela constelação de achados cutâneos característicos, fraqueza muscular e evidências laboratoriais de miosite.
A avaliação laboratorial deve incluir enzimas musculares como CPK e aldolase, além de hemograma, PCR, VHS, testes de função hepática, TSH.
A CPK é mais sensível que a aldolase e é útil no acompanhamento terapêutico. Os níveis de CPK estão elevados cerca de 10 vezes o valor de normalidade.
Também devem ser pesquisados anticorpos antinucleares (FAN) e anticorpos específicos para miosites. Dentre eles, o anti-Jo1 (anti-sintetase) é o único amplamente disponível no nosso meio.
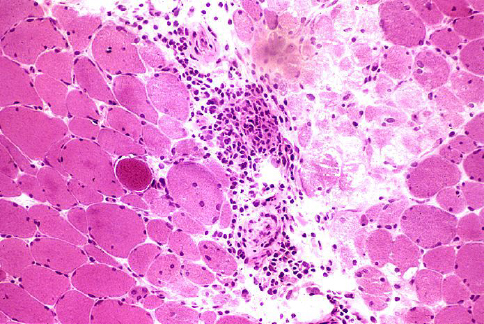
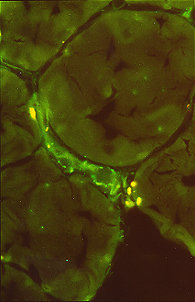
Os achados histopatológicos no DM são variáveis, mas geralmente incluem evidência de lesão de capilares e fibras perifasciculares.
Atrofia perifascicular e fibrose são características da DM, mas podem não ser encontradas em alguns pacientes biopsiados no início da doença.
O complexo de ataque à membrana do complemento terminal C5b-9 é detectável nas paredes dos vasos.
Os critérios diagnósticos usados para as miopatias inflamatórias idiopáticas foram propostos por Bohan e Peter em 1975. São necessários todos os 4 primeiros critérios e para a Dermatomiosite 4 dos 5 critérios, incluindo necessariamente o quadro cutâneo.
Tratamento de dermatomiosite
Os objetivos do tratamento são melhorar a força muscular e evitar o desenvolvimento de complicações extramusculares, além de resolver as manifestações cutâneas na Dermatomiosite.
A principal medicação na terapia inicial é o glicocorticoide. Para pacientes com manifestações cutâneas de DM, recomenda-se corticosteroides tópicos para terapia tópica inicial.
Os inibidores de calcineurina tópicos são uma opção adicional que pode ser útil para o tratamento de longo prazo em áreas da pele com maior risco de atrofia cutânea induzida por corticosteróides.
Para pacientes com miopatia inflamatória associada a fraqueza muscular significativa. recomenda-se glicocorticoides orais.
Normalmente começamos a prednisona com uma dose de 1 mg / kg por dia e geralmente não excedemos 80 mg por dia. Pacientes com apresentações graves podem fazer pulsoterapia com Metilprednisolona (100mg/dia, por 3 dias).
No geral mais de 80% dos pacientes com miopatias inflamatórias melhoram apenas com corticoides. Caso não haja resposta satisfatória, devem ser excluídos os diagnósticos alternativos, miopatia por corticoides e malignidade oculta.
Excluídas essas opções, o próximo passo é a adição de um agente imunossupressor poupador de corticoide, entre eles, azatioprina e metotrexato são os principais representantes.
Conheça nossa pós em dermatologia!
A dermatomiosite, como observamos, é uma doença de ordem reumática com repercussões que envolvem outros sistemas, como a pele e o sistema neurológico.
Sendo assim, o tratamento e acompanhamento multidisciplinar pode ser necessário. Isso irá prover a melhor assistência ao paciente acometido por essa patologia.
Você já conhece o nosso curso de pós-graduação em dermatologia? O programa do nosso curso inclui aulas teóricas e práticas com ambulatório presencial e laboratório de prática profissional.

Sugestão de leitura complementar
Você também pode se interessar por esses artigos:
- Caso Clínico de Neurologia – Dermatomiosite – Sanar Medicina
- Resumo de fibromialgia: fisiopatologia, diagnóstico e tratamento – Sanarflix – Sanar Medicina
- Polimiosite: epidemiologia, fisiopatologia, diagnóstico e tratamento – Sanar Medicina
- Resumo sobre Gota: fisiopatologia, classificação, diagnóstico e tratamento – Sanar Medicina
- Fator antinuclear (FAN): Dicas para a interpretação dos resultados | Colunistas – Sanar Medicina
- Resumo sobre DPFC (Doença por Deposição de Pirofosfato de Cálcio) – Sanar Medicina
Referências bibliográficas
- Clínica Médica – Medicina USP/ HC-FMUSP. Editora Manole. Volume (5), 2009;
- Miller ML, Vleugels RA. Clinical manifestations of dermatomyositis and polymiositis in adults. UpToDate Inc.
- Miller ML, Vleugels RA. Initial treatment of dermatomyositis and polymiositis in adults. UpToDate Inc.
- ORTIGOSA, Luciena Cegatto Martins; REIS, Vitor Manoel Silva dos. Dermatomiosite. An. Bras. Dermatol., Rio de Janeiro , v. 83, n. 3, p. 247-259, June 2008 .
- BRASIL. Ministério da Saúde. Protocolo Clínico e Diretrizes Terapêuticas Dermatomiosite e Polimiosite. Relatório de Recomendação. Brasília: Ministério da Saúde, 2016.
- GIACOMO, C. G. et. al. Atualização em dermatomiosite. Revista Sociedade Brasileira Clínica Médica. São Paulo. Vol. 8, n.5, PP 434-439, set./out. 2010.








