
Miopatias inflamatórias: tudo o que você precisa saber para sua prática clínica!
As miopatias inflamatórias são um grupo heterogêneo de doenças que compartilham a característica comum de lesão muscular imunomediada. Nesse grupo estão tem como principais representantes a dermatomiosite (DM), polimiosite (PM) e miosites por corpúsculos de inclusão (MCI).
Outras doenças menos comuns incluem a síndrome anti-sintetase e a miopatia necrotizante imunomediada (MNI). Muitos pacientes com miopatias inflamatórias não podem ser atribuídos a nenhuma categoria e podem ser classificados como portadores de miosite inespecífica.
Nesse texto iremos comentar principalmente sobre a epidemiologia, patogênese, diagnóstico de tratamento das miopatias inflamatórias mais comuns, a DM, PM e MCI.
Epidemiologia das miopatias inflamatórias
A DM é a miopatia inflamatória mais comum quando consideramos todas as idades. A MCI tem se apresentado como a mais prevalente em pessoas com mais de 50 anos. A incidência anual estimada de DM ou PM varia de dois a dez casos por milhão de habitantes por ano.
A prevalência estimada das MCI em estudo holandês foi de 4,9 por milhão de habitantes e na Austrália de 9,3 por milhão. Como já mencionamos, as outras miopatias inflamatórias idiopáticas são menos comuns.
Qual a patogênese das miopatias inflamatórias?
A DM e PM são miopatias inflamatórias imunologicamente mediadas, caracterizadas por destruição de fibras musculares e infiltração inflamatória dos músculos. Existem diferenças imunológicas e histopatológicas entre os dois distúrbios.
Na dermatomiosite há uma microangiopatia afetando pele e músculo, onde a lise dos capilares endomisiais é causada por ativação e deposição do complemento, levando à isquemia muscular. Entre os pacientes com DM, observou-se superprodução de IFN tipo 1.
A IFN tipo induz a proteína MxA, que é densamente expressa em miofibras perifasciculares e, às vezes, em todas as miofibras.

Na polimiosite, fibras expressando antígenos do complexo principal de histocompatibilidade (MHC) classe I são invadidas por células T citotóxicas CD8-positivas clonalmente expandidas, levando à necrose.
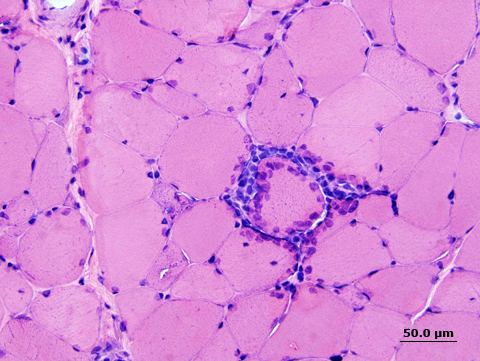
Pacientes com MCI apresentam mecanismo de lesão ainda poucos esclarecidos. Características patológicas distintas são encontradas, incluindo células inflamatórias circundantes e invasoras de miofibras e vacúolos orlados. Algumas miofibras podem ser visivelmente vistas pela microscopia como sendo lesadas por células T citotóxicas.
Quadro clínico das miopatias inflamatórias
Dermatomiosite (DM) e polimiosite (PM) são doenças multissistêmicas com uma ampla variedade de manifestações clínicas. A fraqueza muscular é a característica mais comum de DM e PM.
A maioria dos pacientes exibe fraqueza do músculo esquelético proximal e evidência de inflamação muscular, mas os mecanismos imunológicos e o foco anatômico da lesão dentro do tecido muscular no DM e PM parecem distintos. Pápulas de Gottron e erupção do heliotrópio são aspectos patognomônicos do DM são úteis para distinguir DM de PM.
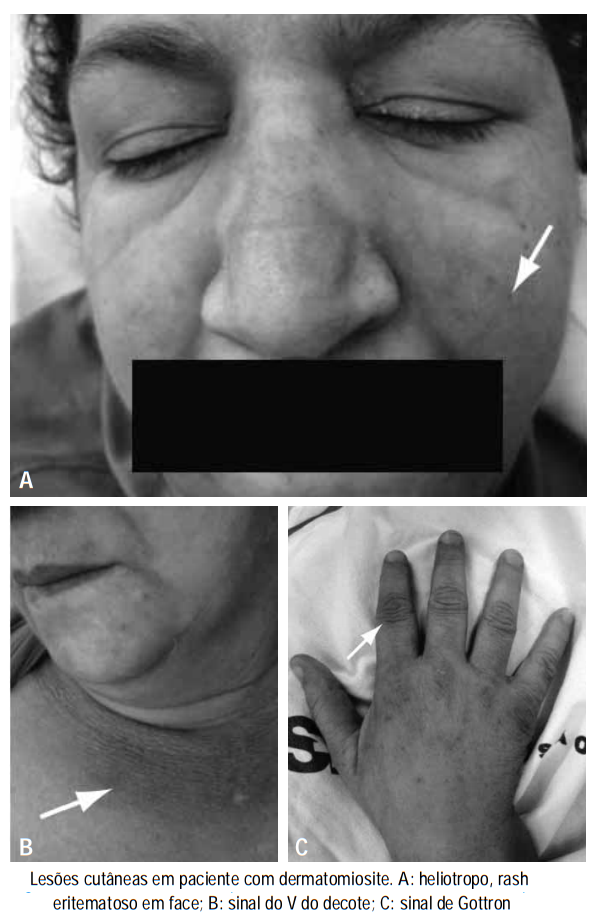
A DM e PM frequentemente estão associadas com dor muscular, sensibilidade dolorosa, disfagia e dificuldades respiratórias. Fenômeno de Raynaud, artralgia, mal-estar, perda de peso e uma febre baixa completam o quadro clínico.
Miosite por corpos de inclusão produz fraqueza nas extremidades inferiores e, depois, nas extremidades superiores. Fraqueza, disfagia e atrofia do quadríceps e dos flexores e extensores do antebraço são características.
A doença é progressiva e está associada com depressão dos reflexos do joelho. Dor muscular ocorre em alguns pacientes e uma fraqueza distal também se desenvolve, mas geralmente é menos grave que a fraqueza proximal.
Como fazer o diagnóstico das miopatias inflamatórias?
O diagnóstico mais preciso de polimiosite ou dermatomiosite se dá através da biópsia muscular com necrose de fibra muscular e infiltração com células inflamatórias.
A CK no soro geralmente está aumentada, às vezes com níveis muito elevados, mas valores normais não excluem o diagnóstico. A eletromiografia (EMG) que apresenta alterações miopáticas e neurogênicas mistas sugere miopatia inflamatória.
Os critérios classificatórios de Bohan e Peter são úteis para auxiliar no diagnóstico, sendo diagnóstico definitivo dado com a presença de 4 critérios, provável se 3 critérios e possível se 2 critérios. Os critérios são:
- Fraqueza muscular proximal e simétrica
- Elevação sérica de enzimas musculares
- Alterações miopáticas na eletromiografia
- Biópsia muscular compatível
- Alterações cutâneas compatíveis com a dermatomiosite.
Na miosite de corpúsculos de inclusão o diagnóstico é confirmado pelo exame histológico do músculo biopsiado. Os níveis séricos de CK podem estar normais ou aumentados e a EMG revela achados inespecíficos, sugestivos de uma miopatia inflamatória.
Diagnósticos diferenciais
lgumas miopatias metabólicas, como a doença de McArdle ou a deficiência de carnitina palmityltransferase (CPT), podem causar fraqueza muscular e dor, mas geralmente não envolvem inflamação muscular.
A polimiosite é uma das principais formas de miopatia inflamatória. Ela envolve inflamação nos músculos, geralmente afetando os músculos proximais (próximos às articulações do ombro e do quadril). No entanto, outras condições podem apresentar características semelhantes, como a dermatomiosite.
A ELA é uma doença neurodegenerativa que pode inicialmente ser confundida com uma miopatia devido à fraqueza muscular progressiva. No entanto, a ELA afeta os neurônios motores, não os músculos diretamente.
Tratamento das miopatias inflamatórias
Na DM e PM o tratamento inicial é feito com fármacos anti-inflamatórios e corticoides. Quando ocorre uma melhora e o valor de CK no soro diminui, a dose de prednisona é gradualmente reduzida para níveis de manutenção.
Imunoglobulina intravenosa é eficaz na dermatomiosite e pode ser usada com os corticosteroides ou no lugar deles. Metotrexato e azatioprina também têm sido usados, seja isoladamente ou em combinação com corticosteróides, em pacientes corticosteróide-resistentes.
A MCI não responde a tratamento imunossupressor ou imunomodulador. O uso de globulina intravenosa tem evidências controversas na doença. Como não há tratamento efetivo, os pacientes podem, eventualmente, permanecer em cadeira de rodas e necessitar de ajuda nas suas atividades diárias.
Dica para aperfeiçoar seu raciocínio neurológico
Conheça nossa pós em neurologia
A neurologia teve um notável crescimento de 111,1% nos últimos 10 anos, conforme revelam os dados da Demografia Médica de 2023. No entanto, esse aumento não ocorreu de maneira uniforme, resultando em escassez de neurologistas em diversas regiões do país e, consequentemente, uma crescente demanda por esses profissionais.
Devido às mudanças no perfil epidemiológico e no estilo de vida da sociedade contemporânea, a necessidade de neurologistas tem aumentado significativamente. O envelhecimento da população mundial é um fator crucial nesse cenário, contribuindo para essa crescente demanda por especialistas na área.
A pós-graduação em neurologia geral é uma ótima possibilidade para os médicos que desejam se capacitar nesta área tão ampla da medicina e que só tem a crescer.
CONHECER PÓS GRADUAÇÃO EM NEUROLOGIA
Sugestão de leitura complementar
- Caso Clínico de Dermatomiosite
- Outras Colagenoses
- Resumo de dermatomiosite
- Resumo sobre DPFC
- Fibromialgia
- Resumo sobre Gota
- Fator antinuclear (FAN): Dicas para a interpretação dos resultados | Colunistas.
- Resumo da Arterite de Takayasu
Referências bibliogáficas
- Tratado de Neurologia da Academia Brasileira de Neurologia. Joaquim Pereira Brasil Neto2o Edição. 2019
- Neurologia Clínica – Greenberg, David A. – Aminoff, Michael J. – Simon, Roger P. 8a Ed. 2014
- REED, Umbertina C.. Doenças neuromusculares. J. Pediatr. (Rio J.) [online]. 2002, vol.78
- KUMMAR, Vina, et. Al. Robbins & Cotran. Bases patológicas das doenças. 8a ed. Rio de Janeiro. Elsevier, 2010.
- Marc L Miller, MD. Diagnosis and differential diagnosis of dermatomyositis and polymyositis in adults. UpToDate, 2020.
- GREENBERG, Steven A. Pathogenesis of inflammatory myopathies. UpToDate, 2020.








